https://doi.org/10.1140/epje/s10189-024-00446-3
Regular Article - Living Systmes
Mathematical modeling for prediction of physicochemical characteristics of cardiovascular drugs via modified reverse degree topological indices
1
Department of Mathematics, Loyola College, 600034, Chennai, India
2
School of Advanced Sciences, Vellore Institute of Technology, 600127, Chennai, India
3
Laboratory of Animal Health Food Hygiene and Quality, University of Ioannina, 47132, Arta, Greece
4
Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, 11461, Riyadh, Saudi Arabia
Received:
7
May
2024
Accepted:
18
July
2024
Published online:
4
August
2024
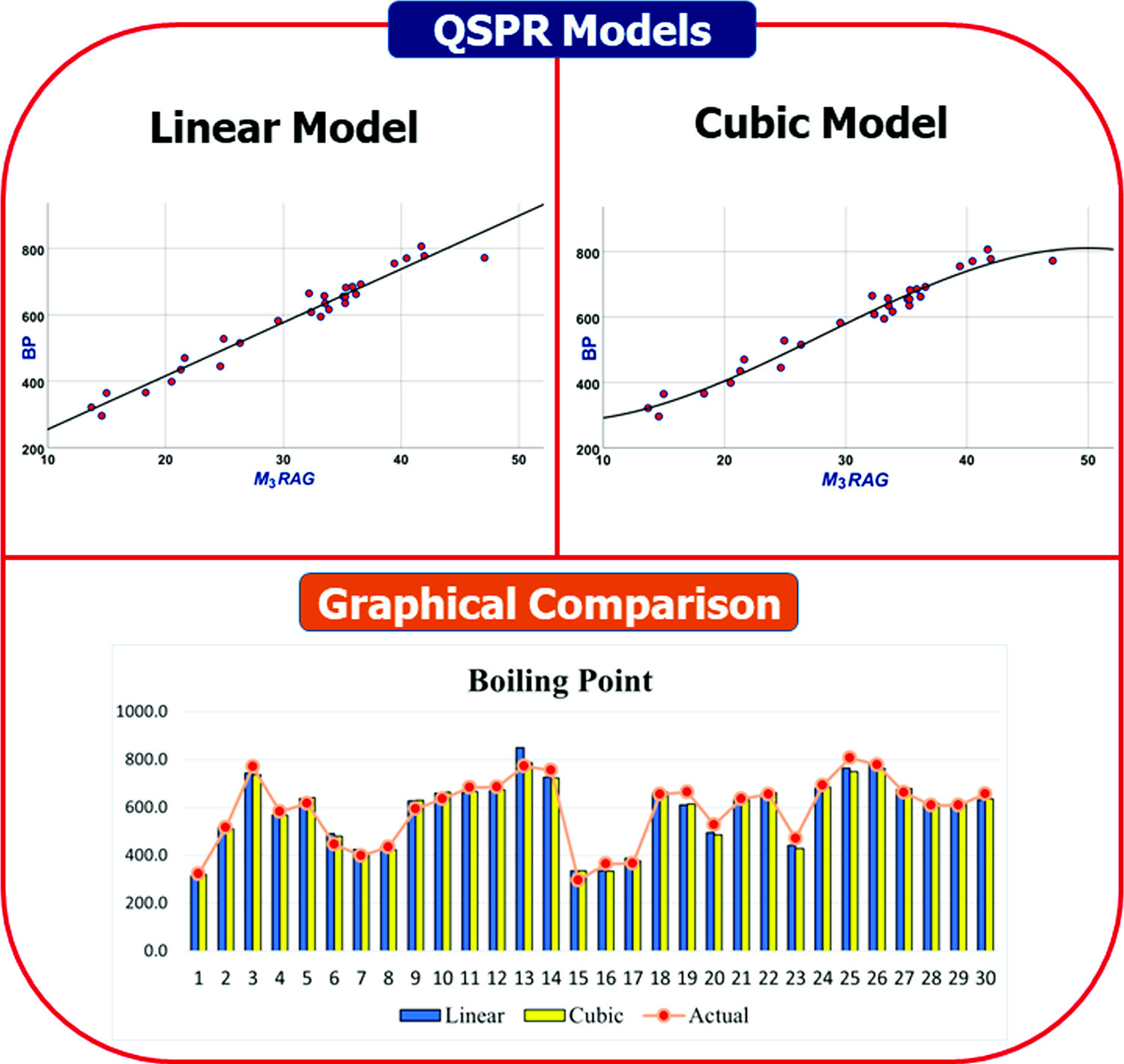
Global health concerns persist due to the multifaceted nature of heart diseases, which include lifestyle choices, genetic predispositions, and emerging post-COVID complications like myocarditis and pericarditis. This broadens the spectrum of cardiovascular ailments to encompass conditions such as coronary artery disease, heart failure, arrhythmias, and valvular disorders. Timely interventions, including lifestyle modifications and regular medications such as antiplatelets, beta-blockers, angiotensin-converting enzyme inhibitors, antiarrhythmics, and vasodilators, are pivotal in managing these conditions. In drug development, topological indices play a critical role, offering cost-effective computational and predictive tools. This study explores modified reverse degree topological indices, highlighting their adjustable parameters that actively shape the degree sequences of molecular drugs. This feature makes the approach suitable for datasets with unique physicochemical properties, distinguishing it from traditional methods that rely on fixed degree approaches. In our investigation, we examine a dataset of 30 drug compounds, including sotagliflozin, dapagliflozin, dobutamine, etc., which are used in the treatment of cardiovascular diseases. Through the structural analysis, we utilize modified reverse degree indices to develop quantitative structure–property relationship (QSPR) models, aiming to unveil essential understandings of their characteristics for drug development. Furthermore, we compare our QSPR models against the degree-based models, clearly demonstrating the superior effectiveness inherent in our proposed method.
Copyright comment Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
© The Author(s), under exclusive licence to EDP Sciences, SIF and Springer-Verlag GmbH Germany, part of Springer Nature 2024. Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.